

Le virus de l'influenza A (IAV) est l'un des pathogènes respiratoires les plus importants dans le porcin. Il a un impact sur la mortalité et provoque des pertes économiques significatives dues à la réduction de la productivité et aux coûts associés aux vaccinations et au traitement. En outre, en raison de la susceptibilité des porcs aux infections par des IAV d'autres espèces, notamment humains, de nouveaux virus peuvent émerger par réarrangement génique qui auraient des implications pour la santé publique. Par conséquent, la compréhension de la diversité génétique des virus circulants dans le porcin permet d'identifier de nouvelles lignées virales et peut constituer un critère sur lequel baser et améliorer les stratégies d'intervention.
La diversité génétique de l'IAV porcin résulte de la dérive et changement antigénique, avec l'introduction de l'IAV d'autres espèces dans les populations porcines (Vincent et coll., 2008). En termes généraux, il existe une co-circulation de trois sous-types dominants (H1N1, H1N2 et H3N2) et quatre lignées génétiques principales décrites en fonction de l'origine des segments géniques. La lignée classique porcine H1 a son origine dans la pandémie de la grippe espagnole de 1918. La deuxième lignée a été détectée à la fin des années 1990 à partir de l'hémagglutinine (HA) et la neuraminidase (NA) dérivées de la grippe humaine saisonnière H3N2. Cette lignée était un nouveau virus réarrangé, contenant des segments géniques HA, NA et PB1 dérivés de la grippe humaine saisonnière H3N2, des segments géniques PB2 et PA d'influenza aviaire et des segments géniques NP, M et NS d'influenza porcine classique H1N1, raison pour laquelle il a été dénommé " virus réarrangé triple ". Ces virus se sont à leur tour réarrangés avec des virus classiques H1N1 pour obtenir l'HA et/ou la NA, ce qui a résulté dans trois nouveaux types génétiques de virus H1N1 et H1N2. La constellation de gènes internes du réarrangement triple (TRIG) est restée relativement cohérente avec les origines porcines (gènes M, NP et NS), aviaires (gènes PB2 et PA) et humaines (PB1) des virus de l'influenza. La troisième lignée s'est produite à partir d'infections répétitives du virus humain saisonnier H1 IAV chez les porcs (ce que l'on appelle " spillover ", ou événement dans lequel un pathogène propre à une espèce saute dans une autre espèce) au début du XXI siècle, ce qui a donné lieu à des lignées différentes de virus humains saisonniers H1N1 et H1N2. La quatrième et plus récente lignée constitue un " spillover " du virus humain saisonnier H3N2 qui a eu lieu en 2010-11. Dans ces quatre lignées principales d'HA, le virus continue à évoluer, en formant de nouveaux clades génétiques (figure 1).

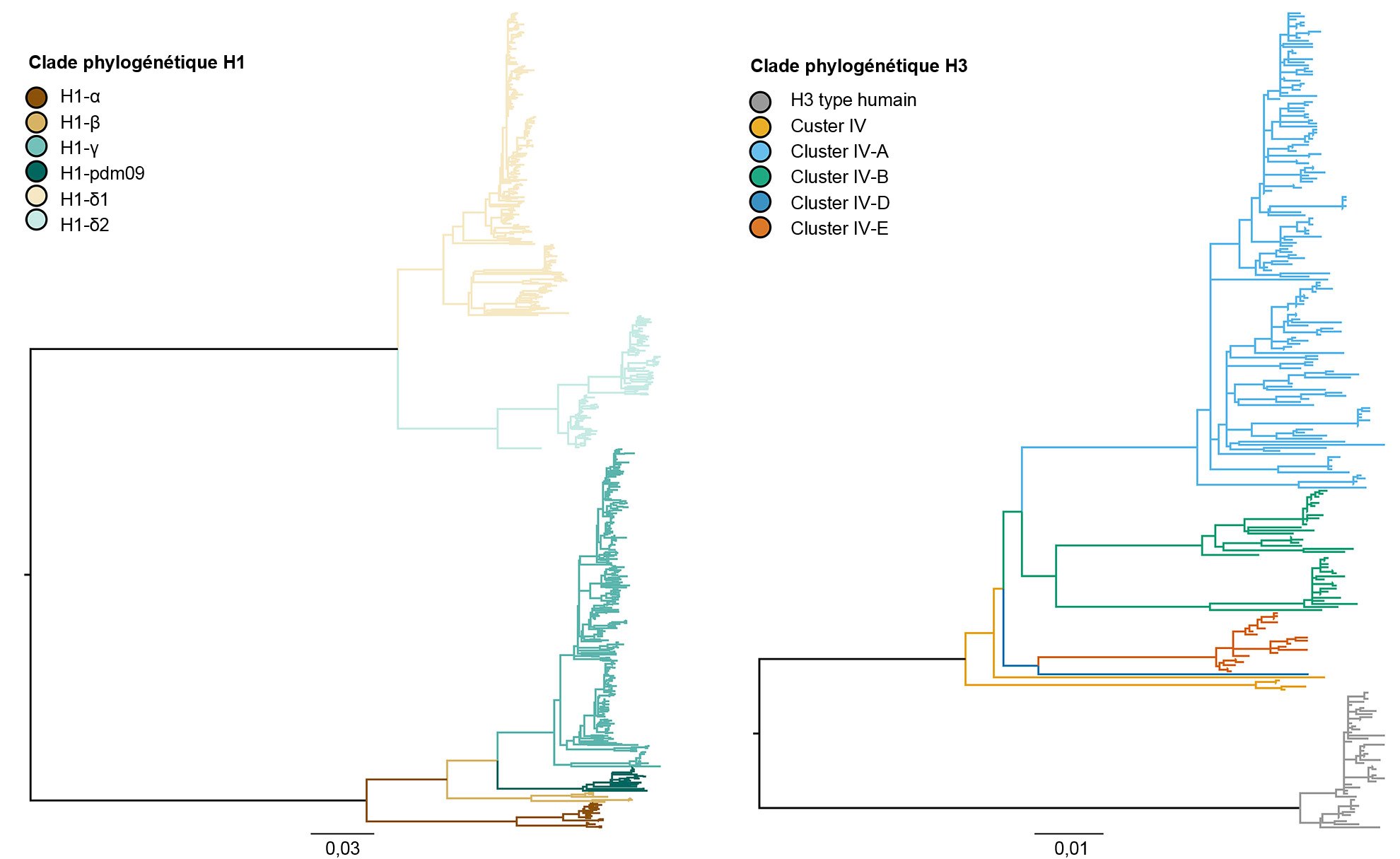
Figure 1. Arbre phylogénétique décrivant les relations génétiques entre des séquences géniques de l'hémagglutinine de l'influenza A porcine H1 et H3 de 2015 générées à partir de méthodes de vraisemblance maximale. Les ramifications en couleur représentent les désignations des clades. La longueur des ramifications est dessinée à l'échelle, et la barre de l'échelle indique le nombre de remplacements de nucléotides par site.
Dans le but d'améliorer la compréhension de la diversité génétique de l'IAV porcin et surveiller le virus pandémique 2009 H1N1 (H1N1pdm09) dans le porcin, en 2009 un système national de surveillance du Département de l'Agriculture des É.-U. (USDA) a été mis en place à travers le réseau national de laboratoires de la santé animale (NAHLN). Le système est basé sur les envois anonymes de producteurs et vétérinaires ; cependant, le système manque d'informations géographiques suffisantes sur la provenance des échantillons. Les données épidémiologiques recueillies comprennent l'état, le type d'échantillon, la raison de l'envoi, l'âge, le type d'emplacement, les résultats de l'analyse et, le cas échéant, les gènes HA, NA et M sont séquencés et déposés dans la base de données de séquences en ligne du Centre National d'Information Biotechnologique, GenBank (Korslund et coll. 2012 ; Anderson et coll. 2013). Grâce à ces données, ainsi qu'aux investissements continus dans le système, l'on dispose d'informations sur les modèles de propagation de l'IAV, leur diversité génétique tout le long de l'année et la dynamique de l'évolution de l'IAV en Amérique du Nord depuis 2010 jusqu'à la date (Anderson et coll., 2013 ; Anderson et coll., 2015 ; Lewis et coll. 2014 ; Rajão et coll. 2015).
Les trois sous-types (H1N1, H1N2 et H3N2) endémiques dans la population porcine des É.-U. ont été détectés tous les ans depuis 2010 jusqu'à nos jours. Les sous-types H1N1 et H1N2 ont été détectés à des fréquences similaires (~35%) pendant ces 5 années. Le sous-type H3N2 représente ~30% des virus identifiés pendant toute la période. Pour décrire la diversité génétique des virus H1, on utilise un système basé sur les lettres de l'alphabet grec pour classer les données des clades d'HA dans les lignées classiques (α, β, γ, γ-2 et H1N1pdm09) ou dans la lignée humaine (δ-1, δ-2). De même, les gènes du cluster IV d'H3 se séparent en 6 clades génétiques (A à F) et un clade H3 type humain (Rajão et coll., 2015 ; Kitikoon et coll. 2013). Cependant, malgré le potentiel de circulation des 14 clades génétiques, la plupart de la diversité d'HA observée dans les exploitations porcines des É.-U. se limite à trois clades. De janvier 2015 à décembre 2015, 43% des isolats H1 appartenaient au clade et à la lignée classique, et 37% ont été classés comme δ-1 dans la lignée humaine saisonnière ; 20% des virus H1 restants appartenaient au clades δ-2, α, H1N1pdm09 et β. Parmi les 8 clades potentiels H3, le cluster IV-A représente la plupart des IAV H3 porcins qui circulent dans les exploitations commerciales (61% des virus H3 en 2015), un nombre croissant d'isolats a été identifié comme H3 type humain (de 5% en décembre 2014 à 25% en décembre 2015), alors que le reste des isolats représente des détections sporadiques des clusters IV-B, IV-C, IV-D, IV-E.
Dans le contexte de l'efficacité des programmes de vaccination, il y a une préoccupation renouvelée sur le fait que la neuraminadase (NA) puisse également jouer un rôle important grâce à sa diversité croissante (Sandbulte et coll., 2016). Même si cela complique la situation, la NA a beaucoup moins de lignées que l'HA. L'HA est couplée aux gènes N2 dérivés d'une ou deux lignées humaines saisonnières H3N2 (qu'elles soient de 1998 ou de 2002 : (Nelson et coll., 2011), aux gènes N1 de la lignée classique porcine, ou aux gènes de la lignée saisonnière humaine H1N1 (Anderson et coll., 2013). Dans l'IAV porcin circulant actuellement, le N1 appartient généralement à la lignée classique (94%) et le N2 à la lignée 2002 (83%), alors que la lignée 1998 représente, même si détectée de manière constante, une petite composante des isolats IAV.
Une deuxième préoccupation concerne les modèles spatiaux dans la diversité de l'IAV porcin. Les États-Unis sont divisés en cinq régions, dans lesquelles les cas sont rapportés sur la base des districts vétérinaires de l'USDA-APHIS (figure 2A). Il existe des différences subtiles et importantes entre les régions par rapport à la diversité et à l'abondance des échantillons fournis au système de surveillance de l'USDA (figure 2B). Alors que dans les régions 1 et 2 on détecte davantage d'H1N1, la région 3 a plus d'H1N2 et la région 4 plus d'H3N2. Même si le cheptel porcin es d'1,6 millions de porcs (USDA-NASS, 2012), il y a très peu de données sur la région 5. La région 2 affiche la plus grande diversité en termes de clades HA et NA différents observés et la plupart des isolats d'IAV porcins provient de cette région. En général, la distribution des clades HA et NA suggère que les décisions régionales ou locales sur les composantes vaccinales peuvent être importantes.

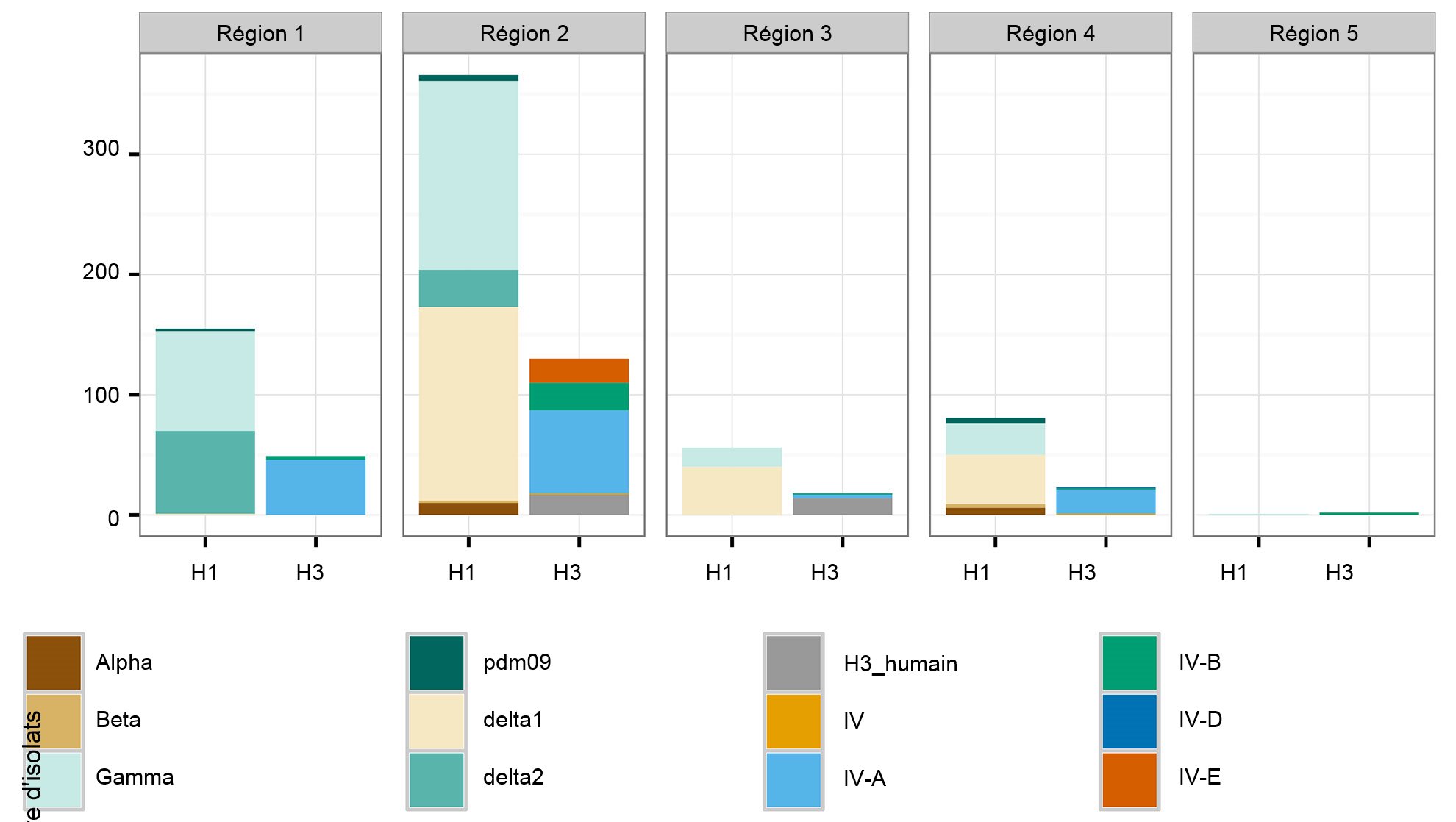
Figure 2. Régions vétérinaires de l'USDA-APHIS (A). Nombre d'isolats d'influenza porcine A recueillis dans chaque région en 2015 et classés selon le clade phylogénétique et colorié comme dans la figure 1 (B).
La diversité génétique de l'IAV porcin est un sujet complexe au niveau régional et particulièrement au niveau global. Aux États-Unis, 17 clades génétiques ont émergé et persisté à partir d'événements de " spillover " entre des hôtes non porcins (c'est à dire, humains) et des processus écologiques et évolutifs subséquents. Même si elle est basée sur différents clades génétiques dérivés de différents épisodes de " spillover " non porcins, la diversité génétique est similaire dans la population porcine mondiale (par exemple, Watson et coll. 2015 ; Bahl et coll. 2015 ; Vijaykrishna D et coll. 2015). La génétique de l'IAV porcin contemporain peut être utilisée pour faire des études sur l'évolution et la diversité antigénique, des travaux qui devraient être réalisés avec des données actuelles fournies par des populations régionales. Cette ensemble de données, avec la mise en place d'une plate-forme vaccinale appropriée, fournirait des données essentielles à utiliser dans la sélection des souches vaccinales ainsi que des informations utiles pour les politiques de gestion des risques pour la santé animale et publique.

